Eigenschaften

Von wenigen Ausnahmen abgesehen gelten für neuromuskuläre Erkrankungen folgende Merkmale:
Es handelt sich meistens um monogene Erbkrankheiten
Die verschiedenen Mendel’schen Erbgänge folgen:
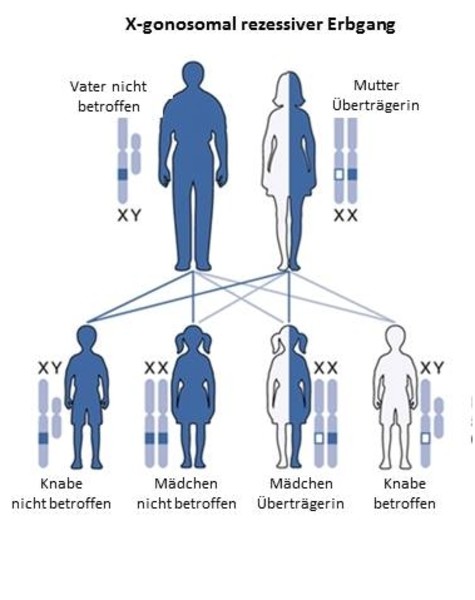
1. X-gonosomal-rezessiver Erbgang (z.B.: Muskeldystrophien Typ Duchenne/Becker)
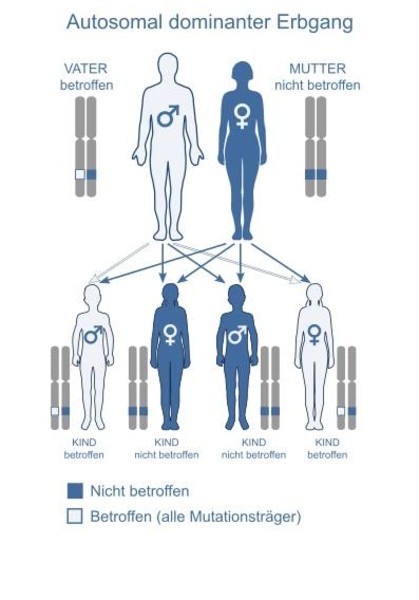
2. autosomal-dominanter Erbgang (Myotone Dystrophie (Myotonie Steinert), Neuropathie Charcot-Marie-Tooth Typ 1A)
3. autosomal-rezessiver Erbgang (z.B. Gliedergürtelmuskeldystrophien, spinal progressive Muskelatrophie)
Sie führen zu einer mehr oder weniger ausgeprägten Invalidität
Zu den typischen Symptomen gehören die Abnahme der Muskelkraft und der Beweglichkeit sowie die zunehmende Versteifung der Gelenke, letztlich – früher oder später - die Unfähigkeit sich selbständig zu bewegen.
Sie sind fortschreitend
Die Verschlimmerung der Symptomatik ist bei den verschiedenen neuromuskulären Erkrankungen unterschiedlich. Einzelne können sich bereits beim Neugeborenen manifestieren (z.B. spinale Muskelatrophie), während andere in der Adoleszenz die Beweglichkeit allmählich einschränken (z.B. Muskeldystrophie) oder erst im Erwachsenenalter ausbrechen und so, wenn auch bedingt, ein normales Leben ermöglichen.
Personen, die weitergehende Information wünschen, wird empfohlen, die Websites der Deutschen Gesellschaft für Muskelkrankheiten (www.dgm.org) oder diejenige von Orphanet zu konsultieren!